PCoA Biplots.
(Look at PCoA for a general overview of PCoA plots!)
To explain the concept of PCoA Biplots, we’ll use the PCoA plot
below:
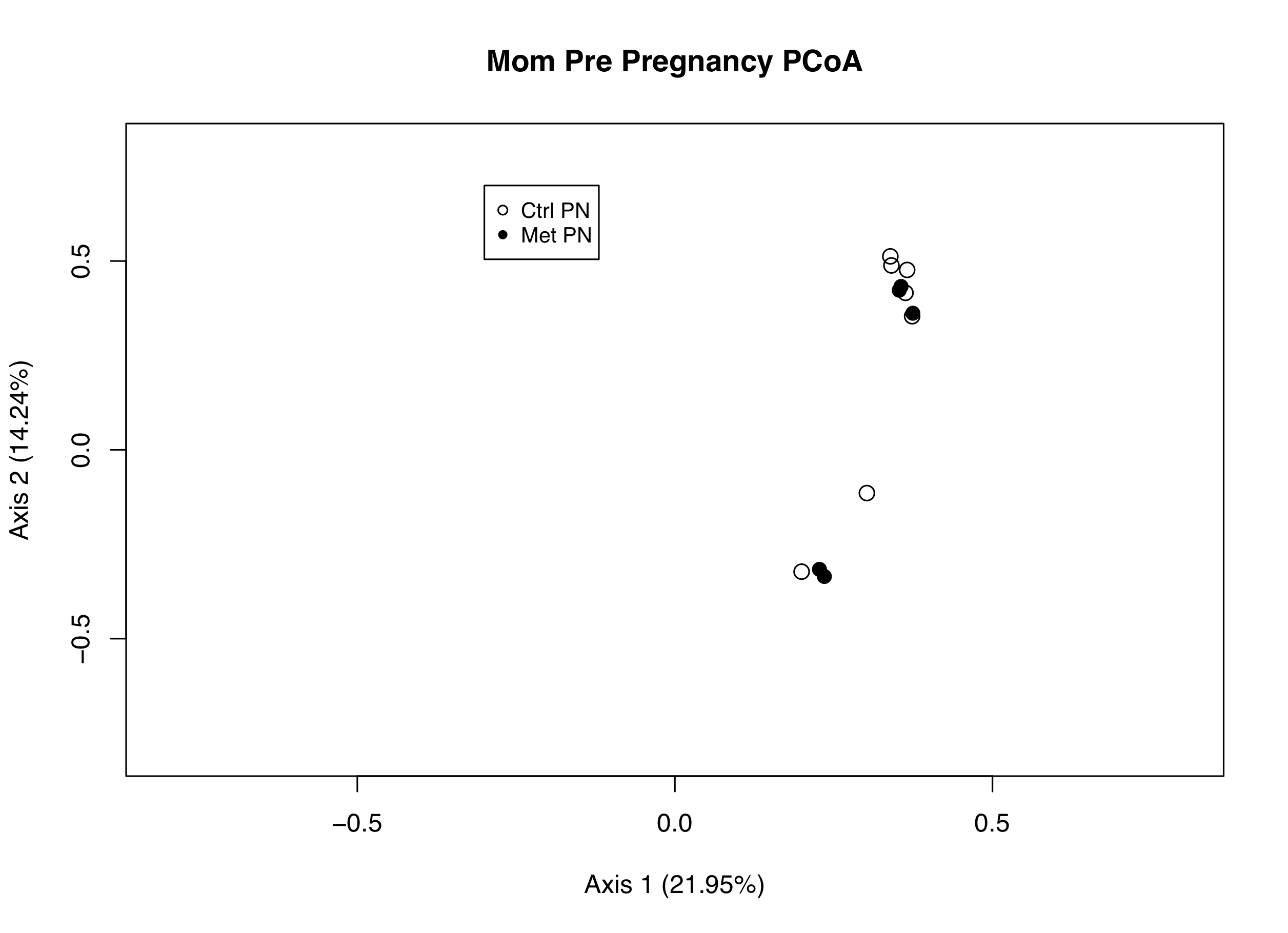
Initially, the PCoA plot suggests the two experimental groups,
Met PN and Ctrl PN, have different microbiomes, considering
the groups do not overlap. The spread within each group seem
similar. Now that we know these microbiomes are different,
our immediate question is: What makes these microbiota different?
To answer this, we can stay within our original PCoA plot.
These microbiomes are different by virtue of the most significant
OTUs observed. In other words, whatever OTUs differ the most
amongst groups is what’s causing these microbiomes to not overlap.
To see which OTUs are involved we can add biplot arrows.
By displaying these biplot arrows we can see the OTUs
represented as vectors. These vectors show which direction
any given OTU is pushing the cluster(s).
To better explain this, let’s see some biplot arrows in action!
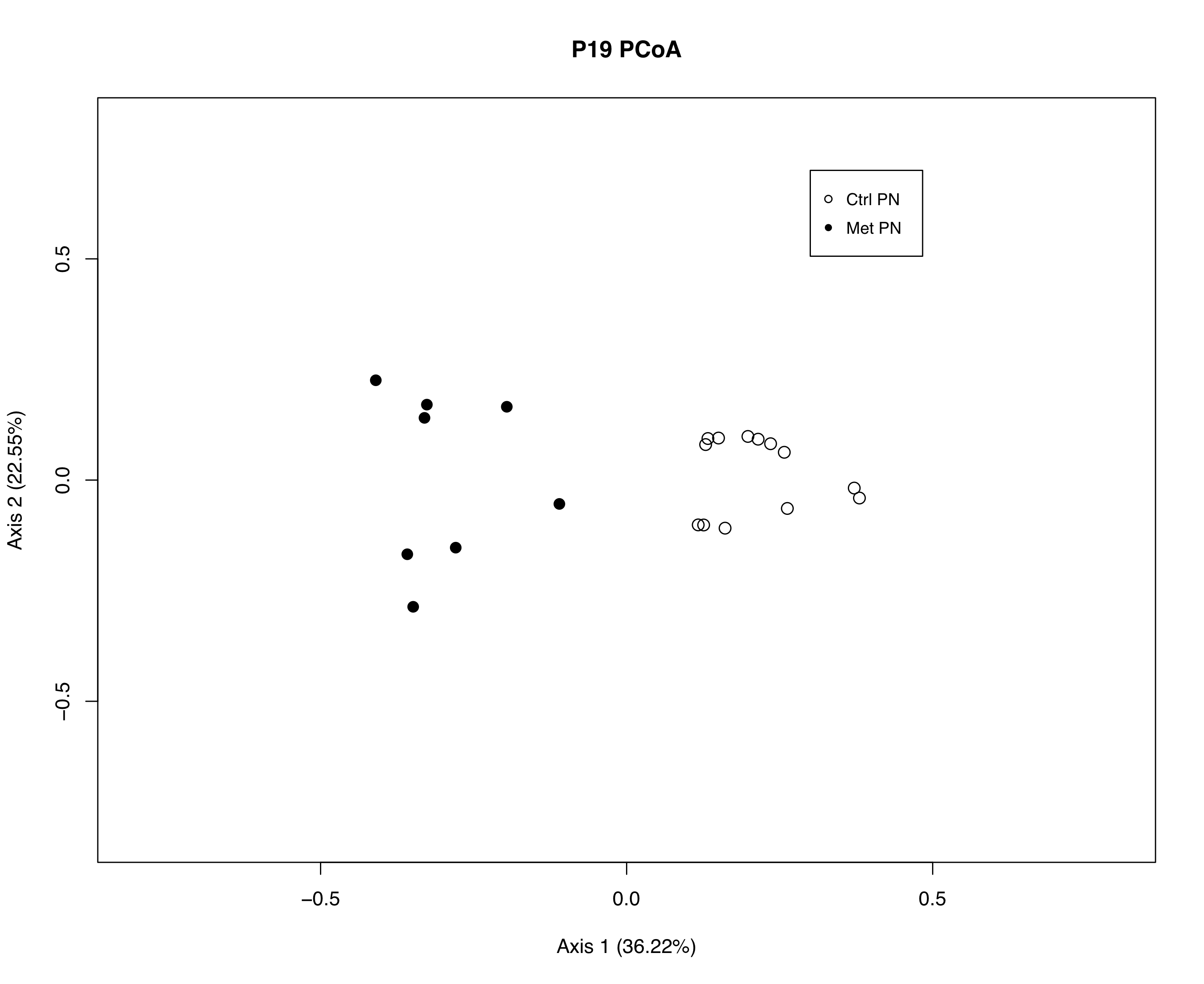
Let’s dig deeper into the above plot and see which OTUs are
pushing Met PN and Ctrl PN to diverge. (Note: for sanity
purposes, the PCoA and PCoA Biplot app are separate, so the
plots will have different formatting) Take a look at the
PCoA Biplot below:
While this plot looks different than our bare PCoA plot, it’s
more similar than it may appear. Both experimental groups are
the same, Met PN and Ctrl PN. Each experimental group has the
same shape (Met PN has filled circle, Ctrl PN has open circle).
As stated above, the plots look different. This is because of
two reasons that are specific to our coding. First, our PCoA
and PCoA Biplot Shiny Apps are built separately, so the
formatting isn’t exactly the same. Additionally, this plot
example contains 6 additional Ctrl PN microbiomes (the 6 dots
clustering at the bottom of the plot).
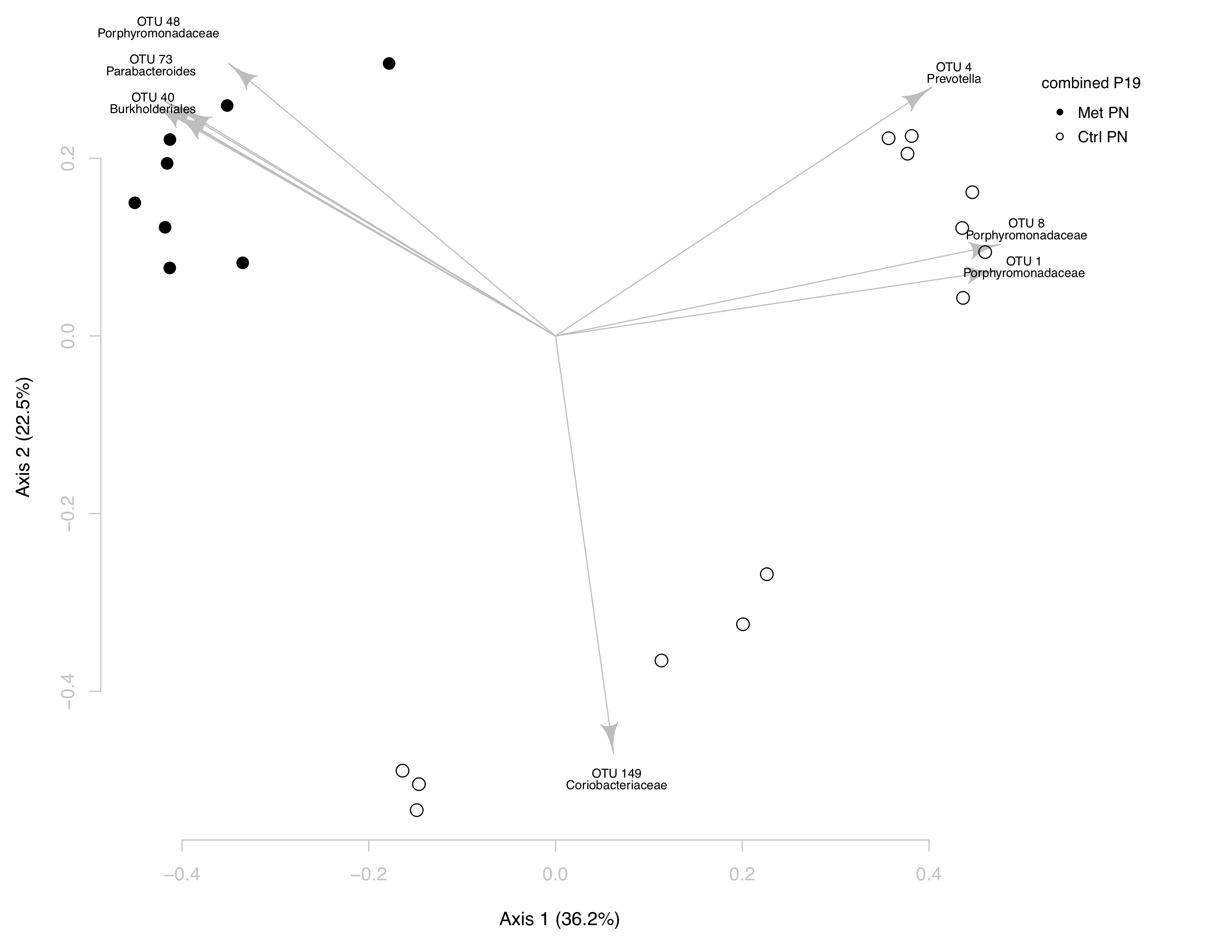
Now that we know what’s similar and different between the two
plots, let’s start exploring our biplot! While it’s apparent
the groups have different microbiomes in the bare plot, this
biplot shows which OTUs are creating that difference.
In the upper left hand corner of the plot, it’s clear the
Met PN samples are being changed by three OTUs:
OTU 48, OTU 73, and OTU 40.
These OTUs correspond to the genuses Porphyromonadaceae,
Parabacteroides, and Burkholderiales, respectively.
When addressing the OTUs that are changing Ctrl PN relative to
Met PN, there are two distinct shifts. First, is the cluster
of Ctrl PN at the bottom of the plot, which is shifted by
OTU 149, Coriobacteriaceae. Second, is the
cluster of Ctrl PN at the upper right hand corner of
the plot, which is shifted by three OTUs: OTU 1,
OTU 4, and OTU 8. These OTUs correspond
to the phyla Prevotella, Porphyromonadaceae, and
Porphyromonoadaceae, respectively.
A hypothetical next step in data analysis would be to identify
the points in Ctrl PN. It seems that within
Ctrl PN, the microbiomes are separating into two
distinct groups. Is this due to a protocol change? Are these
two groups different cohorts of the same experiment? Are the
two groups separating based on gender? The possibilities are endless!


![Thumbnail [100%x225]](images/cards/microbiome.png)
![Thumbnail [100%x225]](images/cards/abundance.png)
![Thumbnail [100%x225]](images/cards/pcoa.png)
![Thumbnail [100%x225]](images/cards/pcoa-bi.png)
![Thumbnail [100%x225]](images/cards/bf.png)
![Thumbnail [100%x225]](images/cards/lda.png)